Лекция № 7. Закономерности управления простым обратимым гомогенным процессом
Обратимыми называются процессы, в которых одновременно протекают прямая и обратная реакции.
Рассмотрим реакцию aA + bB ↔ cC + dD.
В начальный момент времени скорость прямой реакции, которая пропорциональна произведению концентраций реагентов А и В
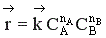 ,
,
больше скорости обратной реакции, пропорциональной произведению концентраций продуктов С и D
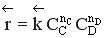 .
.
Но с течением времени, по мере увеличения концентрации веществ C и D скорость обратной реакции увеличивается, а скорость прямой реакции уменьшается. В некоторый момент времени они становятся равными 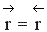 ; наступает химическое равновесие. После достижения равновесия соотношение участников реакции остается постоянным во времени. Это соотношение называется константой равновесия.
; наступает химическое равновесие. После достижения равновесия соотношение участников реакции остается постоянным во времени. Это соотношение называется константой равновесия.
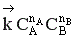 =
=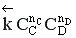
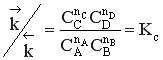
Для газофазных реакций константу равновесия можно выразить через парциальные давления участников реакции, которые пропорциональны их концентрации pi = Ci RT.
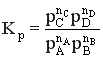
Между этими константами существует связь 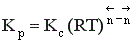 , где
, где 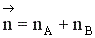 - суммарный порядок прямой реакции,
- суммарный порядок прямой реакции, 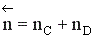 - суммарный порядок обратной реакции.
- суммарный порядок обратной реакции.
Константу равновесия газофазной реакции можно также выразить через мольные доли компонентов
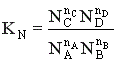 .
.
Парциальное давление каждого компонента пропорционально мольной доле его в смеси pi = p Ni, где р – общее давление системы, 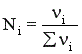 , νi – число молей данного компонента. Тогда
, νi – число молей данного компонента. Тогда 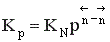 .
.
Константы Кс и Кр зависят только от температуры, а константа КN еще и от давления.
В случае реальных газовых смесей и растворов концентрации и парциальные давления в выражении констант меняют на активность «а» и фугитивность «f».
Итак, пределом протекания обратимых процессов является состояние равновесия, при котором скорости прямой и обратной реакции равны, а концентрации и парциальные давления продуктов и реагентов остаются постоянными во времени. При этом достигается некоторая максимальная в этих условиях степень превращения реагентов – равновесная конверсия α* и равновесный выход целевого продукта -β*. Их величина зависит от степени смещения равновесия в сторону образования целевого продукта. Следовательно, при управлении обратимым процессом важно не только обеспечить высокую скорость процесса, но и создать условия, при которых химическое равновесие смещено в сторону образования целевого продукта.
Характеристика модели:
1) Процесс простой – можно описать одним стехиометрическим уравнением аА + bВ ↔ cС + dD, где С – целевой продукт;
β = α, так как весь реагент превращается только в целевой продукт.
2) Процесс обратимый – αmax = α*≠ 100%.
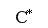
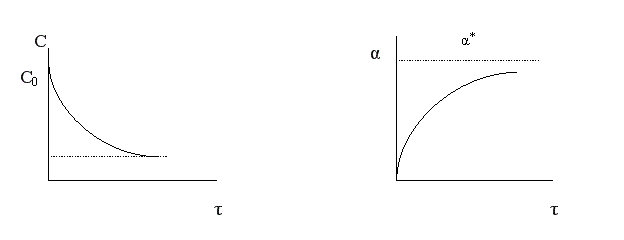
-
Процесс гомогенный – скорость процесса равна скорости химической реакции. Однако следует помнить, что в обратимых процессах скорость равна разности скорости прямой и обратной реакции
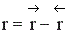 . Она уменьшается во времени и при
. Она уменьшается во времени и при 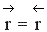 (в момент наступления химического равновесия) равна нулю.
(в момент наступления химического равновесия) равна нулю.
Объекты управления: скорость химической реакции и положение равновесия.
Инструменты управления: кинетические параметры.
Правила смещения равновесия в обратимых процессах сформулированы химиком Ле-Шателье и состоят в следующем:
«Если на систему, находящуюся в равновесии, воздействовать извне, изменяя какое-нибудь из условий, определяющих равновесие, то равновесие смещается в том направлении, в котором эффект воздействия уменьшается».
Смещение равновесия происходит только в том случае, когда произведенное воздействие неодинаково влияет на скорость прямой и обратной реакции. Например, катализатор всегда одинаково ускоряет как прямую, так и обратную реакцию, и поэтому не оказывает влияния на положение равновесия.
Рассмотрим, как влияет на скорость и положение равновесия изменение таких технологических параметров, как температура, давление, концентрация реагентов.
-
Влияние температуры
При повышении температуры скорость химической реакции увеличивается за счет увеличения константы скорости, причем, чем больше энергия активации процесса, тем больше влияние температуры на скорость. Энергия активации эндотермического процесса всегда больше энергии активации обратного экзотермического процесса на величину теплового эффекта реакции. Покажем это на энергетической диаграмме.
По оси «y» отложим энергию рассматриваемой системы, а по оси «x» - путь или координату реакции. Пусть реакция взаимодействия А+В является экзотермической, ∆Н < 0. Тогда энергия системы в результате реакции будет понижаться; энергетический уровень продуктов С и D будет меньше энергетического уровня исходных реагентов. Разница между энергиями конечного и начального состояния есть тепловой эффект процесса ∆Н.
Чтобы молекулы веществ А и В прореагировали, они должны преодолеть некоторый энергетический барьер. На это затрачивается энергия, называемая энергией активации 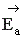 . Для того, чтобы прореагировали вещества C и D, они тоже должны обладать энергией активации
. Для того, чтобы прореагировали вещества C и D, они тоже должны обладать энергией активации 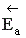 .
.
Как видно из рисунка, энергия активации обратной реакции больше энергии активации прямой реакции < 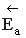 .
.
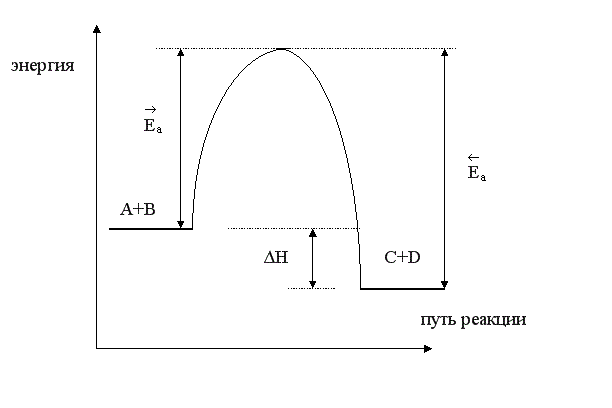
Если реакция взаимодействия А + В будет эндотермической, то получим такую же диаграмму, отличающуюся лишь тем, что энергетический уровень системы А + В ниже, чем уровень энергии системы C + D. Энергия активация прямой реакции будет выше энергии активации обратной реакции > .
Таким образом, энергия активации эндотермического процесса всегда больше энергии активации обратного экзотермического процесса на величину теплового эффекта реакции. Следовательно, повышение температуры увеличивает в большей степени скорость эндотермической реакции.
Если реакция образования целевого продукта С происходит с выделением тепла, повышение температуры увеличит в большей степени константу скорости обратной реакции, чем константу скорости прямой реакции; константа равновесия уменьшится, равновесие сместится в сторону образования исходных веществ А и В.
Если ∆Н < 0, при Т↑ 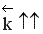 ,
, 
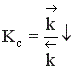
Если прямая реакция эндотермическая, повышение температуры в большей степени увеличит константу прямой реакции; константа равновесия увеличится, равновесие сместится в сторону образования целевого продукта.
Если ∆Н > 0, при Т↑ 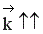 ,
, 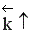 ,
, 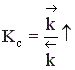 .
.
Смещение равновесия означает изменение величины равновесной конверсии и выхода; в случае простого процесса они синхронно уменьшаются при увеличении температуры в экзотермических процессах и увеличиваются в эндотермических.
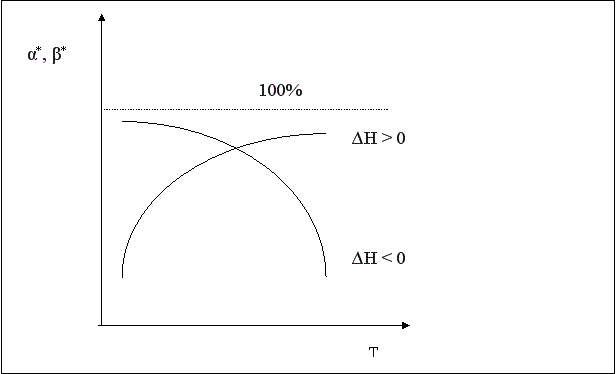
Степень смещения равновесия при изменении температуры зависит от величины теплового эффекта. Чем больше ∆Н, то есть чем больше разница энергий активации прямой и обратной реакции, тем больше смещается равновесие. Если ∆Н ≈ 0, температура почти не влияет на положение равновесия.
Задача управления обратимым процессом заключается в создании условий, при которых равновесие смещено в сторону образования целевого продукта с одновременным обеспечением достаточно высокой скорости достижения равновесия.
Пусть протекает простая необратимая реакция типа 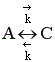 .
.
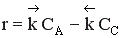
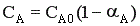
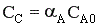
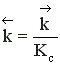
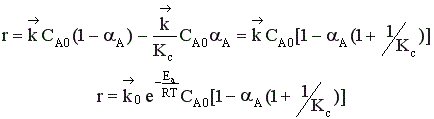
Из уравнения видно, что для некоторой постоянной конверсии αА скорость процесса при повышении температуры должна возрастать за счет увеличения члена 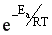 , а с другой стороны, может уменьшаться, если уменьшается константа равновесия Кс, или увеличиваться, если Кс увеличивается.
, а с другой стороны, может уменьшаться, если уменьшается константа равновесия Кс, или увеличиваться, если Кс увеличивается.
При Т↑ ↑ r↑;
если Кс ↓ r↓, если Кс ↑ r↑
Первый случай реализуется в экзотермических реакциях. В обратимых экзотермических реакциях при повышении температуры скорость сначала возрастает за счет увеличения , достигает некоторого максимального значения, а затем начинает уменьшаться за счет уменьшения константы равновесия.
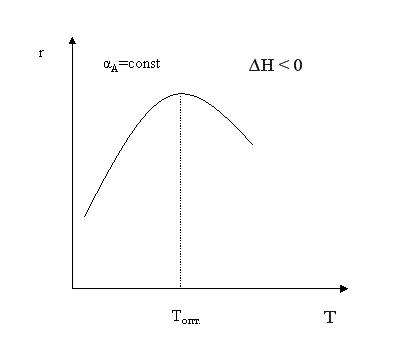
В области низких температур при повышении температуры скорость процесса увеличивается за счет увеличения скорости прямой экзотермической реакции; обратная эндотермическая реакция при этих температурах еще практически не протекает. При переходе к более высоким температурам начинает протекать обратная реакция, ее скорость в большей степени возрастает под воздействием температуры, чем скорость экзотермической реакции. В результате суммарная скорость процесса начинает уменьшаться. Температура, при которой достигается максимальная скорость, называется оптимальной.
Из уравнения также видно, что при увеличении конверсии αА скорость процесса уменьшается.
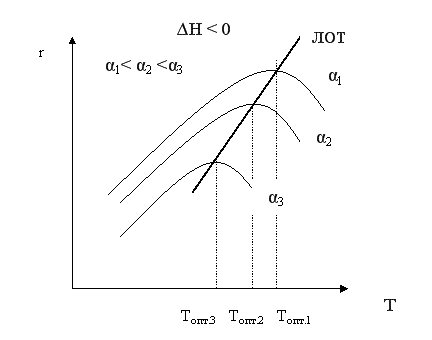
Если α2 > α1, кривая зависимости r =f (T) располагается ниже кривой, соответствующей α1. Если α3 > α2, то соответствующая кривая располагается еще ниже и т.д. Это связано с тем, что для того, чтобы достичь более высоких значений конверсии, требуется больше времени (максимальная конверсия достигается в момент наступления равновесия). Но по мере приближения к равновесию скорость процесса падает и в момент равновесия она равна нулю. Для каждой величины конверсии получается своя кривая зависимости r =f (T), причем, чем выше конверсия, тем ниже расположена эта кривая и тем меньше оптимальная температура. Линия, соединяющая максимумы полученных таким образом кривых, является линией оптимальных температур (ЛОТ). Эта линия показывает, как нужно изменять температуру в ходе процесса для того, чтобы увеличить степень превращения реагентов, работая все время при максимально возможных в этих условиях скоростях процесса.
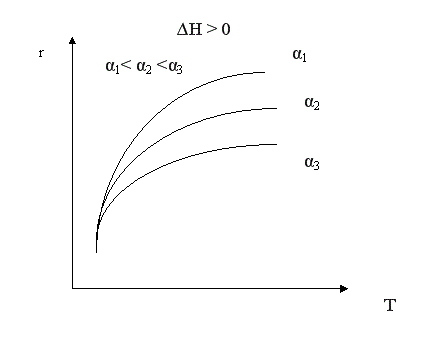
В простых обратимых эндотермических процессах скорость процесса при повышении температуры возрастает и за счет увеличения члена , и за счет увеличения Кс. При увеличении конверсии (при прочих равных условиях) скорость процесса уменьшается.
Установим зависимость конверсии от температуры α = f (T) для тех же реакций.
Для обратимого экзотермического процесса зависимость α = f (T) при τ = const сначала увеличивается, достигая максимального значения, а затем снижается. Снижение αА связано с достижением равновесной конверсии, которая при повышении температуры в экзотермическом процессе уменьшается за счет смещения равновесия.
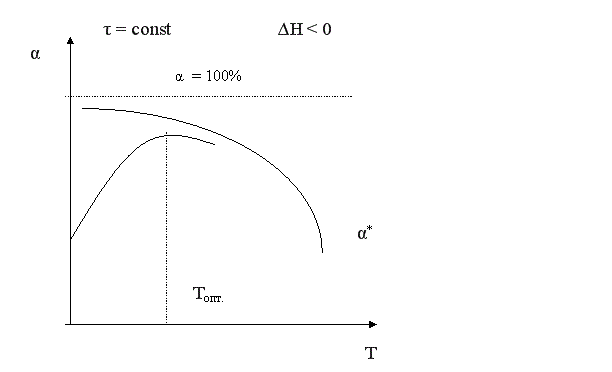
Кривая, соответствующая τ2 > τ1, располагается выше, чем τ3 > τ2 – еще выше и т.д. Кривая, соединяющая максимумы, является линией оптимальных температур (ЛОТ).
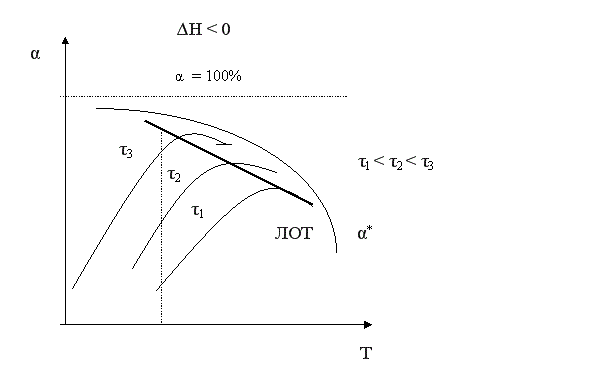
Таким образом, для обратимого экзотермического процесса не существует единой оптимальной температуры. В начале процесса нужно задавать более высокую температуру для поддержания высокой скорости процесса, а затем температуру следует снижать по ЛОТ, чтобы при достижении более высоких значений конверсии поддерживать максимально возможную скорость процесса.
Для простого обратимого эндотермического процесса вопрос о влиянии температуры решается однозначно: при повышении температуры увеличивается и равновесная и фактическая степень превращения. Однако, и в этом случае есть некоторая оптимальная температура, выше которой процесс вести нерационально, поскольку конверсия увеличивается незначительно.
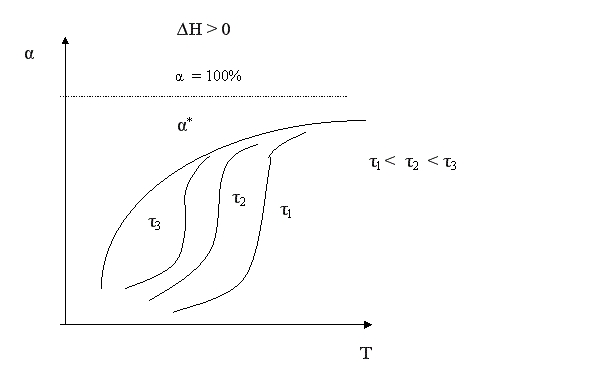
Для всех кривых зависимости α = f(Т) характер изменения конверсии при невысоких температурах одинаков, поскольку в этих условиях влияние термодинамических факторов (т.е. обратной реакции) незначительно. С повышением температуры влияние термодинамических факторов увеличивается. Степень этого влияния зависит от типа реакции. Для необратимых реакций кривые зависимости α = f(Т) асимптотически приближаются к единице. Для обратимых реакций они ограничиваются кривой равновесной степени превращения, которая для экзотермических процессов с повышением температуры уменьшается, а для эндотермических процессов возрастает.
-
Влияние концентрации участников реакции
Суммарная скорость обратимого процесса равна
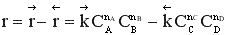
При увеличении начальной концентрации исходных реагентов А и В увеличиваются их текущие концентрации, а, следовательно, скорость прямой реакции и скорость всего процесса в целом (химическое равновесие устанавливается быстрее).
Константа равновесия 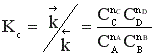 при этом не меняется: во сколько раз увеличивается произведение равновесных концентраций реагентов, во столько раз увеличивается и произведение равновесных концентраций продуктов. Однако равновесный состав смеси будет другой. Покажем это на примере образования йодоводорода.
при этом не меняется: во сколько раз увеличивается произведение равновесных концентраций реагентов, во столько раз увеличивается и произведение равновесных концентраций продуктов. Однако равновесный состав смеси будет другой. Покажем это на примере образования йодоводорода.
Н2 + I2 ↔ 2HI; 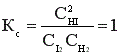
Пусть С0 (I2) = 1 моль/л; С0 (Н2) = 1 моль/л.
К моменту установления равновесия образовалось х моль HI, при этом в реакцию вступило х/2 моль I2 и Н2. Равновесные концентрации I2 и Н2 равны (1 – 0,5х).
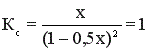 ; 0,75 х2 + х – 1 = 0; х = 0,7
; 0,75 х2 + х – 1 = 0; х = 0,7
Состав равновесной смеси: 0,7 моль/л HI, (1-0,7/2) =0,65 моль/л Н2 и I2.
Равновесная конверсия водорода равна 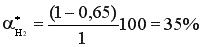 .
.
Увеличим начальную концентрацию йода:
С0 (I2) = 2 моль/л; С0 (Н2) = 1 моль/л.
Равновесная смесь содержит х моль HI, (2-0,5) моль I2 и (1-0,5х) моль Н2.
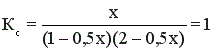 ; 0,75х2 + 1,5х – 2 = 0; х = 0,9
; 0,75х2 + 1,5х – 2 = 0; х = 0,9
Состав равновесной смеси: 0,9 моль/л HI, (2-0,9/2) = 1,55 моль/л I2; (1-0,9/2) = 0,55 моль/л Н2.
Равновесная конверсия водорода равна 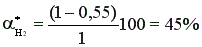 .
.
При увеличении концентрации одного из реагентов увеличивается равновесная конверсия другого реагента, выход целевого продукта увеличивается.
Таким образом, в случае обратимого процесса также целесообразно работать с высококонцентрированным сырьем и можно использовать избыток одного из реагентов. Это позволяет не только поддерживать высокую скорость процесса в течение необходимого времени, но и сдвигает равновесие в сторону образования целевого продукта.
Равновесие обратимого процесса можно сместить вправо также уменьшая концентрацию продуктов С и D, выводя их из реакционной зоны. Константа равновесия Кс при этом также не изменяется: во сколько раз уменьшается произведение концентраций продуктов, во столько же раз уменьшается и равновесная концентрация исходных реагентов. При заданных значениях начальных концентраций реагентов это означает увеличение степени переработки сырья.
Уменьшение концентрации продуктов в реакционной смеси приводит к уменьшению скорости обратной реакции и к увеличению суммарной скорости процесса.
Способы удаления продуктов из зоны реакции могут быть разные. Продукт либо химически связывают вводимым извне веществом, например,
СО + Н2О ↔ Н2 + СО2 СО2 + СаО → СаСО3↓,
либо переводят в другое фазовое состояние (конденсируют или кристаллизуют при охлаждении, испаряют или возгоняют при нагревании и т.д.). В последнем случае, как правило, используют циркуляционную схему процесса.
-
Влияние давления
Скорость обратимого газофазного процесса
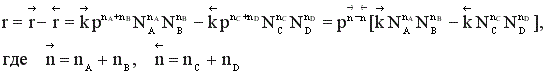
зависит от давления, причем тем больше, чем больше отличаются порядки прямой и обратной реакции.
Если реакция протекает без изменения числа молей, скорость обратимого процесса не зависит от давления. Если число молей в результате реакции уменьшается, увеличение давления приводит к увеличению скорости процесса. В противном случае повышение давления вызывает уменьшение скорости установления химического равновесия.
при р↑ r = const, если 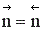 ;
;
r↑, если 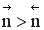
r↓, если 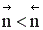
Давление также влияет на положение равновесия. Для анализа этого влияния воспользуемся уравнением 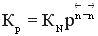 .
.
Константа равновесия КР зависит только от температуры, поэтому при Т = const, КР = const.
Если , КР = КN = const. Константа КN не зависит от давления; давление не влияет на положение равновесия, на равновесную конверсию и выход.
Если , увеличение давления приводит к уменьшению 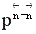 , КN должна увеличиться, равновесие сдвигается в сторону образования целевого продукта.
, КN должна увеличиться, равновесие сдвигается в сторону образования целевого продукта.
Если , увеличение давления приводит к увеличению , КN уменьшается, равновесие сдвигается в сторону образования исходных реагентов.
Таким образом, если число молей в результате реакции уменьшается, увеличение давления способствует сдвигу равновесия в сторону образования целевого продукта и увеличению скорости процесса. Если число молей в результате прямой реакции увеличивается, для увеличения скорости процесса и сдвига равновесия в сторону образования целевого продукта нужно уменьшить давление в системе. Если 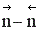 близко к нулю, использовать давление для повышения эффективности процесса нецелесообразно, так как затраты намного превысят полученный эффект.
близко к нулю, использовать давление для повышения эффективности процесса нецелесообразно, так как затраты намного превысят полученный эффект.
Обобщая вышесказанное, можно сделать выводы, что
-
для эффективного проведения простого обратимого гомогенного процесса необходимо не только обеспечить высокую скорость процесса, но и сместить равновесие в сторону образования целевого продукта;
-
равновесие можно смещать, используя такие технологические параметры, как температура, давление, концентрация. Выбор метода смещения определяется расчетами.
-
Для обратимого экзотермического процесса требования кинетики и термодинамики находятся в противоречии, поэтому регулирование температуры необходимо осуществлять по линии оптимальных температур.
-
Повышение давления благоприятно лишь для газофазных процессов, протекающих с уменьшением числа молей. Но и в этом случае существует некоторое оптимальное давление, выше которого работать нецелесообразно из-за экономических соображений.
-
В обратимых процессах желательно осуществлять ступенчатое удаление продуктов реакции из реакционной зоны, а также работать с избытком одного из реагентов, причем вводить его можно тоже ступенчато.
-
В случае, когда условия, благоприятствующие смещению равновесия в сторону образования целевого продукта, приводят к уменьшению скорости процесса, в начале поддерживают высокую скорость, а затем создают условия для получения максимально возможного равновесного выхода.
-
Чтобы сохранить достаточно высокую скорость обратимого процесса (производительность реактора), процесс осуществляют до степени превращения реагентов 0,8 – 0,9 от равновесного значения.
Пример: Технология получения серной кислоты. Окисление сернистого газа в серный ангидрид.
2SO2 + O2 ↔ 2SO3, ∆Н = -94,23 кДж/моль
Реакция окисления сернистого газа характеризуется очень высоким значением энергии активации, поэтому ее практическое осуществление возможно лишь в присутствии катализатора – V2O5.
Технологическая классификация процесса: простой, обратимый, каталитический, газофазный, но гетерогенный, экзотермический.
Объекты управления: скорость процесса и положение равновесия.
В промышленности для окисления сернистого газа используют ванадиевые контактные массы БАВ, СВД, СВС, ИК, в составе которых ~8% V2O5, нанесенного на пористый носитель. Катализатор очень чувствителен к контактным ядам, поэтому обжиговый газ перед подачей на окисление тщательно очищают от пыли, соединений мышьяка и фтора.
При окислении концентрированных газовых смесей лимитирующей может стать перенос вещества из газового потока к поверхности катализатора (внешняя диффузия). При работе на обычных концентрациях сернистого газа (5-8%) лимитирующей стадией процесса является проникновение реагирующих веществ в поры катализатора к внутренней поверхности зерен (внутренняя диффузия). Поэтому внутренняя поверхность зерен не используется полностью, особенно на начальных стадиях процесса. С ростом температуры степень использования внутренней поверхности уменьшается. При высоких степенях превращения, температуре 4700С и коротких порах процесс переходит в кинетическую область протекания.
Влияние температуры. Так как реакция окисления SO2 является обратимой экзотермической реакцией, температурный режим ее проведения должен приближаться к линии оптимальных температур. На выбор температурного режима дополнительно накладываются два ограничения, связанные со свойствами катализатора. Нижним температурным пределом является температура зажигания ванадиевых катализаторов (400 - 4400 С); верхний предел определяется температурой, при которой происходит перестройка структуры катализатора и потеря его активности (600 – 6500 С). В диапазоне 400 – 600 0С процесс стремятся провести так, чтобы по мере увеличения степени превращения температура уменьшалась. Чаще всего в промышленности используют полочные контактные аппараты с наружным теплообменом. Схема теплообмена предполагает максимальное использование теплоты реакции для подогрева исходного газа и одновременное охлаждение газа между полками.
Влияние состава исходной смеси. Скорость реакции повышается с ростом концентрации кислорода, поэтому процесс проводят при его избытке (обычно 3-х кратный по сравнению со стехиометрией). Для этого более концентрированный обжиговый газ (14-15% SO2) разбавляют воздухом перед стадией контактного окисления.
Одной из важнейших задач в производстве серной кислоты является увеличение степени превращения сернистого газа и снижение его выбросов в атмосферу. Эта задача может быть решена несколькими методами. Самым рациональным из них является метод двойного контактирования и двойной адсорбции. Его сущность состоит в том, что реакционную смесь, в которой степень превращения SO2 составляет 90-95%, охлаждают и направляют в промежуточный абсорбер для выделения SO3. В оставшемся реакционном газе соотношение О2 : SO2 существенно повышается, что приводит к смещению равновесия реакции вправо. Вновь нагретый реакционный газ снова подают в контактный аппарат, где на одном – двух слоях катализатора достигают 95%-ной степени превращения оставшегося сернистого газа. Суммарная степень превращения SO2 составляет в таком процессе 99,5 – 99,8%.
Абсорбция триокисда серы. Для полного извлечения SO3 в качестве сорбента используют 98,3%-ную серную кислоту (техническое название – моногидрат), соответствующую азеотропному составу. Использование в качестве поглотителя менее концентрированной кислоты и тем более воды приводит к образованию сернокислотного тумана, плохо улавливаемого в обычной абсорбционной аппаратуре. При использовании 100%-ной серной кислоты или олеума в паровой фазе велико равновесное парциальное давление SO3, поэтому он абсорбируется не полностью.
Для снижения равновесное давление паров серной кислоты поддерживают температуру не выше 1000С.
Технологическая схема производства серной кислоты из колчедана методом двойного контактирования.
Колчедан через дозатор подают в печь КС. Полученный запыленный обжиговый газ, содержащий 13% SO2 и имеющий температуру 7000С, подают сначала в котел-утилизатор, а затем на стадию сухой очистки от пыли. В котле–утилизаторе происходит охлаждение газа с одновременным получением энергетического водяного пара (давление 4 МПа, температура 4500С).
В очистном отделении, состоящим из двух промывных башен, двух пар мокрых электрофильтров и сушильной башни происходит очистка газа от соединений мышьяка, селена, фтора и его осушка. Перед сушильной башней обжиговый газ разбавляют воздухом. После сушильной башни обжиговый газ проходит через фильтр-брызгоуловитель и поступает в турбогазодувку, где нагревается продуктами реакции до температуры зажигания катализатора 420-4400С. Затем он поступает на первый слой контактного аппарата. В первом слое катализатора происходит окисление сернистого газа на 74% с одновременным повышением температуры до 6000С. После охлаждения до 4650С газ поступает на второй слой контактного аппарата, где степень превращения достигает 86%. А температура газа возрастает до 5140С. После охлаждения до 4500С газ поступает на третий слой контактного аппарата, где степень превращения увеличивается до 94-94,5%, а температура повышается до 4700С.
Затем в соответствии с требованиями метода двойного контактирования и двойной абсорбции реакционный газ охлаждают в теплообменниках до 1000С и направляют на абсорбцию первой ступени. После абсорберов газ нагревают до температуры 4300С и подают на четвертый слой катализатора. В четверном слое происходит окисление диоксида серы на 80%, сопровождающееся повышением температуры до 4490С. Реакционную смесь вновь охлаждают до температуры 4090С и направляют на пятый последний слой контактного аппарата. Общая степень превращения после пяти стадий контактирования составляет 99,95. Газовую смесь после охлаждения направляют в абсорбер второй ступени. Непоглощенный газ, состоящий в основном из воздуха, очищают и выбрасывают в атмосферу.